1. Introduction
Microbiome research stands at the frontier of biological discovery, reshaping our understanding of how microbial communities influence ecosystems, human health, agriculture, and global biogeochemical cycles. These microscopic assemblages — from the ocean’s depths to the human gut — are responsible for fundamental processes such as nutrient cycling, disease resistance, and host development. Marine microbial communities alone account for more than 80 % of Earth’s biomass and form the base of global food webs, emphasizing the enormity of their ecological influence (Azam & Worden, 2004). However, the very tools that have propelled microbiome science forward — high-throughput DNA sequencing and culture-independent methods — also harbor systematic and random biases that can distort biological interpretations at every step of the analytical workflow, from study design to data interpretation, threatening reproducibility and comparability across studies.
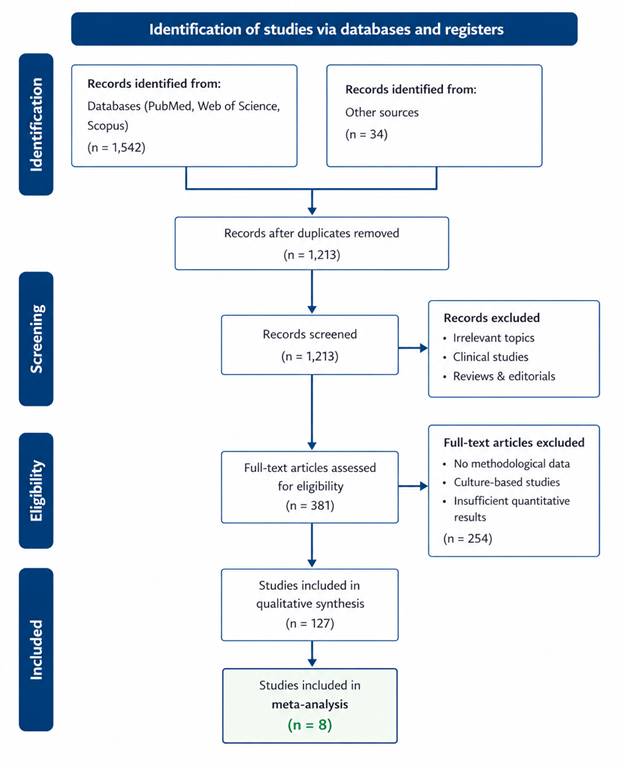
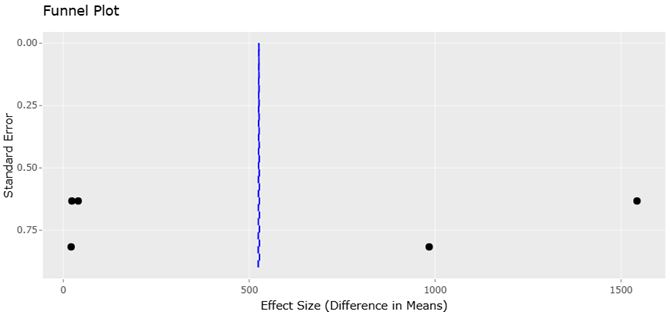
This review is grounded in a systematic examination of methodological biases that pervade microbiome research and a meta-analytic synthesis of how these biases affect observed outcomes such as species richness and community structure. The goal is to illuminate the sources of bias and provide a coherent narrative that helps researchers recognize how seemingly innocuous decisions — from sample collection techniques to bioinformatic pipelines — can yield radically different biological conclusions.
At the conceptual level, bias in microbiome research behaves like a sieve with variable mesh sizes: if the mesh is too coarse, it misses rare taxa that may be biologically important; if the mesh is uneven, it skews the apparent composition of communities (Francioli et al., 2021). The first critical stage of microbiome investigations — experimental design — sets the stage for everything that follows. thoughtful consideration of ecological context, relevant covariates, and appropriate controls are essential to reduce environmental confounds and technical contamination (Costea et al., 2017). Failure to incorporate standardized metadata such as diet, age, or environmental conditions can obscure true biological patterns and magnify false associations.
Sampling methodology introduces one of the most pervasive forms of bias. In low-biomass environments like the skin, the choice between swabs, biopsies, tape stripping, or specialized microprojection arrays determines not only the microbial biomass collected but also the depth and diversity of organisms sampled (Bjerre et al., 2019; Meisel et al., 2016; Santiago-Rodriguez et al., 2023). Surface swabs tend to capture transient or superficial taxa and are particularly sensitive to “kitome” contamination from reagents and extraction kits, while deeper biopsies can access anaerobic bacteria residing in follicles and glands. The influence of these methods extends beyond superficial differences; it fundamentally alters the perceptible community structure, reinforcing the need for niche-specific standardized protocols.
Following collection, DNA extraction stands as another critical bottleneck. Many kits rely on mechanical lysis techniques such as bead beating to rupture rigid cell walls, yet the efficacy of lysis varies widely among microbial taxa. Some commercial platforms may robustly extract DNA from Gram-negative bacteria while underrepresenting Gram-positive cells with thicker peptidoglycan layers, leading to bias in downstream analyses (Shi et al., 2022). Choosing an extraction method therefore requires not only technical awareness but also a clear understanding of the ecological questions at hand.
The choice of genetic markers and primer design arguably exerts the greatest influence on perceived community composition. Universal primers targeting regions like the 16S rRNA gene promise broad coverage but often perform unevenly across taxa. For instance, in silico analyses have shown that certain “universal” primer sets fail entirely to amplify key genera such as Bifidobacterium, introducing false absences into gut microbiota profiles (Mancabelli et al., 2020). Similarly, comparative studies in oral microbiome research demonstrate that targeting the V1-V2 region yields superior species-level resolution for Streptococcus compared to commonly used V3-V4 primers, which struggle to distinguish closely related taxa (Na et al., 2023). These primer biases are not mere technical footnotes; they shape our fundamental interpretation of microbial ecology and disease associations.
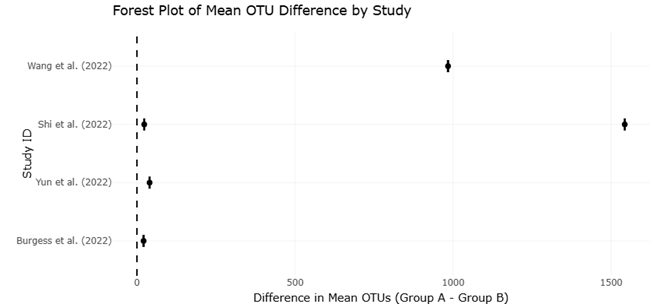
Beyond primer choice, the variable region of the 16S rRNA gene targeted for amplification heavily skews taxonomic visibility. Studies have shown that different regions, such as V1-V2 versus V3-V4, capture different subsets of bacterial diversity, often underestimating richness when suboptimal regions are used (Klindworth et al., 2013). Traditional short-read sequencing platforms like Illumina provide high-throughput data but are constrained to partial gene sequences. In contrast, third-generation long-read technologies such as PacBio sequencing can generate full-length 16S rRNA gene sequences, offering finer taxonomic resolution and revealing a greater number of operational taxonomic units (OTUs) — a pattern that has been consistently borne out in complex environmental samples like marine biofilms (Wang et al., 2022).
Further complicating matters are technical artifacts such as polymerase chain reaction (PCR) bias, chimeric sequence generation, and the influence of intergenomic phenomena like mitochondrial heteroplasmy. In some taxa, inherent genetic complexity may lead to “wrong species delimitation with high confidence,” where standard barcodes and primers preferentially amplify one genotype over another, masking true biological diversity (Martínez et al., 2023). These molecular biases underscore the limitations of relying on single genetic markers like cytochrome c oxidase subunit I (COI) for comprehensive biodiversity assessments (Folmer et al., 1994; Hebert et al., 2003).
The subsequent analytical phase — bioinformatics processing — introduces its own set of interpretative choices. Early microbiome studies often clustered sequences into OTUs based on arbitrary similarity thresholds (typically 97 %), a practice that masks fine-scale variation. The advent of amplicon sequence variant (ASV) methods represents a methodological leap by distinguishing sequences down to single-nucleotide differences, improving both resolution and reproducibility (Callahan et al., 2017; Eren et al., 2015). However, the accuracy of taxonomic assignments remains dependent on reference databases such as SILVA, Greengenes, and UNITE, which vary in completeness and curation quality (Balvočiūtė & Huson, 2017; Quast et al., 2013).
Addressing these biases is not just a matter of technical optimization; it is central to the integrity of microbiome science. Standardization initiatives such as the Minimum Information about any Sequence (MIxS) guidelines strive to harmonize reporting across studies, enabling meta-analyses that compare findings across ecosystems, hosts, and experimental designs (Francioli et al., 2021). Moreover, rigorous contamination control measures — including extraction blanks, mock community standards, and randomized processing blocks — are essential for validating biological signals against reagent-derived noise.
The importance of these methodological considerations is vividly illustrated in case studies spanning diverse environments. For example, marine biofilms analyzed using full-length 16S sequencing consistently reveal higher species richness than those assessed by partial gene fragments (Wang et al., 2022). Similarly, investigation into freshwater sediments demonstrates that extraction kits may yield comparable prokaryotic riches but differ significantly in eukaryotic recovery, illuminating taxon-specific extraction biases (Shi et al., 2022). In model organisms like mice, diet — a strong biological covariate — has been shown to exert a greater influence on gut community structure than exercise, emphasizing the need to integrate biological context into methodological interpretation (Yun et al., 2022).
In summary, the field of microbiome research has matured rapidly, yet this progress has not eradicated the methodological pitfalls that can distort scientific conclusions. From sampling and extraction to marker choice and analytical pipelines, each decision carries the potential to either illuminate or obscure biological reality. By systematically reviewing these biases and synthesizing their impacts through meta-analysis, this review aims to equip researchers with a nuanced understanding of the methodological landscape. Only by recognizing and rigorously controlling for bias can the promise of microbiome science be fully realized — transforming raw sequence data into meaningful insights about life’s invisible majority.