3.1 Fermentation as Cultural Memory and Biological Innovation
Traditional Chinese fermented foods represent far more than culinary artifacts preserved through history; they are dynamic biological systems shaped by centuries of environmental adaptation, empirical experimentation, and cultural continuity. Long before the emergence of microbiology as a scientific discipline, communities across China were already manipulating microbial processes to preserve food, enhance flavor, and support health. Archaeological findings from Jiahu suggest that fermented beverages derived from rice, honey, and fruits were produced as early as 7000 BC, making fermentation one of humanity’s earliest forms of biotechnology (Ray & Joshi, 2014). In many ways, these early fermentation practices reflected necessity—food preservation in unstable climates, seasonal scarcity, and the need to transform raw agricultural products into more digestible forms. Yet over time, fermentation evolved into something more sophisticated and culturally embedded.
Traditional fermented products such as Baijiu, soy sauce, sufu, douchi, paocai, and fermented rice wines became deeply integrated into regional identity and dietary customs. Their production was rarely standardized in the modern industrial sense. Instead, fermentation depended on local climate, indigenous microbial populations, artisanal knowledge, and inherited sensory judgment. This variability, once viewed as inconsistency, is now increasingly recognized as part of the ecological richness that defines traditional fermentation systems. What contemporary science is beginning to uncover is that these foods harbor remarkably complex microbial ecosystems whose biological interactions shape not only flavor and texture but also nutritional and therapeutic potential.
Recent advances in microbiome science have further complicated our understanding of fermentation. The microbial communities within traditional Chinese fermented foods are not static assemblages of isolated organisms. Rather, they are evolving ecological networks characterized by cooperation, competition, metabolic exchange, and environmental adaptation. Zhang et al. (2019) argued that the microbial ecology of traditional Chinese fermented foods should be approached as a systems-level phenomenon rather than a collection of independent fermentative strains. This perspective has become increasingly important as researchers attempt to bridge traditional fermentation knowledge with modern omics technologies and precision food engineering. Figure 1 summarizes the conceptual transition of traditional Chinese fermentation from ancient culturally inherited practices to contemporary microbiome-driven food innovation, emphasizing the ecological complexity and systems-level interactions within fermented food ecosystems.
3.2 The Philosophical and Biomedical Intersection: Food as Medicine
One of the more intriguing aspects of traditional Chinese fermented foods is the longstanding belief that food and medicine are fundamentally interconnected. Within traditional Chinese medicine (TCM), the spleen–stomach system is often described as the “foundation of postnatal existence,” responsible for transforming food into qi, blood, and bodily nourishment. Fermented foods such as Suan Cai, Dou Chi, and Jiu Niang have historically been consumed not simply for taste but also for digestive regulation, immune support, and restoration of internal balance.
For a long time, these interpretations were largely regarded through philosophical or symbolic frameworks. However, contemporary microbiome research has begun to identify possible physiological mechanisms underlying some of these traditional observations. Lim et al. (2025) suggested that fermented foods may function as mediators between ancient Eastern dietary wisdom and modern gut microbiota science. What traditional medicine conceptualized as “digestive imbalance” or “stagnant qi” increasingly resembles what biomedical science now describes as microbial dysbiosis. Alterations in gut microbial composition have been associated with obesity, inflammatory bowel disease, metabolic syndrome, depression, and immune dysfunction, and fermented
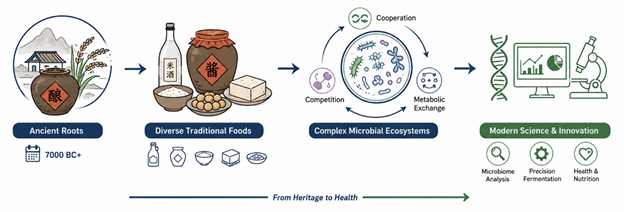
Figure 1: Traditional Chinese Fermentation as Cultural Memory and Biological Innovation. This figure illustrates the historical evolution and systems-level complexity of traditional Chinese fermented foods, beginning from ancient fermentation practices and progressing toward modern microbiome science, precision fermentation, and health-oriented food innovation. The figure highlights the interconnected roles of cultural heritage, indigenous microbial ecology, metabolic exchange, and emerging omics technologies in shaping the nutritional, sensory, and therapeutic properties of fermented food systems.
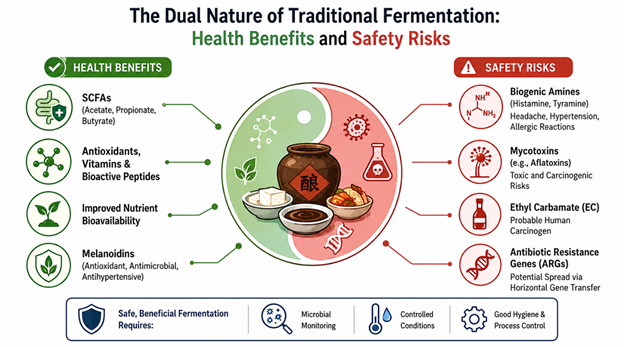
Figure 2: The Dual Nature of Traditional Fermentation: Balancing Functional Health Benefits and Microbial Safety Risks. This figure summarizes the beneficial effects of traditional fermented foods, including SCFA production, antioxidant activity, and improved nutrient bioavailability, alongside major safety concerns such as biogenic amines, mycotoxins, ethyl carbamate, and antibiotic resistance genes. The figure highlights the importance of microbial monitoring and controlled fermentation practices for maintaining safe and functional fermentation ecosystems
foods may help modulate these microbial environments through probiotics, prebiotic substrates, and microbial metabolites.
This does not necessarily mean that traditional theories directly predicted modern microbiome science. The relationship is likely more nuanced and indirect. Nevertheless, the overlap is difficult to ignore. Fermented foods contain bioactive peptides, organic acids, vitamins, enzymes, and microbial metabolites that may influence gut barrier integrity, inflammatory signaling, and metabolic regulation (Xing et al., 2023). Increasingly, researchers are exploring whether traditional dietary patterns rich in fermented products contributed historically to microbial resilience and metabolic adaptability in East Asian populations.
3.3 The Microbial Architecture of Traditional Fermentation
At the center of traditional Chinese fermentation lies the fermentation starter, commonly known as Qu or Koji. Unlike industrial fermentation systems that rely on monocultures or highly controlled starter inocula, traditional starters are biologically heterogeneous and environmentally derived. They contain diverse assemblages of molds, yeasts, and bacteria that collectively initiate and sustain fermentation processes (Yang et al., 2022).
Molds, particularly filamentous fungi such as Aspergillus, Rhizopus, and Mucor, play foundational roles during early fermentation stages. These organisms produce hydrolytic enzymes—including amylases, cellulases, and proteases—that degrade complex carbohydrates and proteins into fermentable substrates. In soy sauce and rice wine production, Aspergillus oryzae is especially important because it catalyzes starch saccharification and amino acid release, creating substrates that support downstream microbial succession (Nout & Aidoo, 2010).
Yeasts contribute primarily through alcoholic fermentation and aroma development. Species such as Saccharomyces cerevisiae metabolize sugars into ethanol while simultaneously generating volatile esters, aldehydes, and aromatic alcohols that define the sensory complexity of products like Huangjiu and Baijiu (Chen et al., 2020). However, yeasts rarely function independently. Their metabolic activity often depends on nutrient availability generated by molds and bacteria earlier in fermentation.
Lactic acid bacteria (LAB), including Lactobacillus plantarum, Pediococcus, and Leuconostoc species, contribute acidification, pathogen suppression, and flavor stabilization (Li et al., 2023). Through lactic acid production and bacteriocin synthesis, LAB establish environmental conditions that inhibit spoilage organisms while extending product shelf life. In fermented vegetables such as paocai and suansun, LAB-driven acidification also shapes characteristic sourness and textural changes (Hu et al., 2021).
What is increasingly evident, however, is that these organisms operate not as isolated agents but as members of highly interactive ecological communities. Fan et al. (2023) demonstrated that synergistic microbial fermentation improves both safety and sensory quality through cooperative metabolic relationships. Nutrient cross-feeding, environmental modification, and signaling interactions appear essential for maintaining fermentation stability. Consequently, removing organisms from their ecological context may oversimplify their functional significance.
3.4 From Culture-Based Microbiology to the Omics Era
Historically, fermentation microbiology depended heavily on culture-based techniques. Researchers isolated microorganisms using agar media, characterized colony morphology, and identified strains through biochemical testing. While these methods remain useful for industrial applications, they capture only a fraction of microbial diversity present in traditional fermented foods.
Many microorganisms enter viable but non-culturable (VBNC) states during fermentation because of environmental stressors such as ethanol accumulation, osmotic pressure, nutrient limitation, or acidic conditions. Although these organisms remain metabolically active, they fail to grow under standard laboratory conditions (Wang et al., 2023). As a result, substantial microbial diversity remained effectively invisible for decades.
The development of next-generation sequencing (NGS) and multi-omics technologies fundamentally changed this situation. Metagenomics now allows direct sequencing of microbial DNA from food matrices without prior cultivation, enabling researchers to identify both dominant and low-abundance taxa simultaneously. Metaproteomics and metabolomics extend this approach by revealing active protein expression and metabolite production during fermentation (Yang et al., 2020).
These technologies have revealed that uncultured microorganisms often contribute significantly to flavor biosynthesis and metabolic regulation. In Shaoxing-jiu fermentation, metagenomic analysis demonstrated that microbial diversity in JIUYAO starters strongly influences the formation of volatile compounds and fermentation dynamics (Chen et al., 2020). Similarly, shotgun metagenomic analysis of suansun identified carbohydrate and amino acid metabolism as key drivers of flavor development (Hu et al., 2021). Such findings suggest that fermentation outcomes depend not merely on microbial presence but on coordinated metabolic interactions occurring across entire microbial ecosystems.
3.5 The Dual Nature of Traditional Fermentation: Health Benefits and Safety Risks
Traditional fermented foods are often celebrated for their probiotic and nutritional benefits, yet their biological complexity also introduces important safety concerns. This tension has been described as “two sides of the same coin” (Xu et al., 2022). Fermentation can simultaneously generate beneficial metabolites and hazardous compounds depending on microbial composition, environmental conditions, and process control.
On the beneficial side, fermented foods are important sources of short-chain fatty acids (SCFAs), antioxidants, vitamins, and bioactive peptides. SCFAs such as acetate, propionate, and butyrate contribute to gut barrier integrity, immune modulation, and inflammatory regulation (Xing et al., 2023). Fermentation may also improve nutrient bioavailability and reduce anti-nutritional compounds in plant-based foods. Melanoidins, formed during Maillard reactions in products such as soy sauce and vinegar, represent another biologically active component. Yang, Fan, and Xu (2022) reported that melanoidins exhibit antioxidant, antimicrobial, and antihypertensive activities, suggesting that fermentation-associated browning reactions may contribute functional health benefits beyond flavor enhancement. Nevertheless, uncontrolled fermentation conditions may also facilitate the production of hazardous metabolites. Biogenic amines (BAs), including histamine and tyramine, are among the most significant concerns in protein-rich fermented foods. These compounds arise primarily through microbial amino acid decarboxylation and may trigger headaches, hypertension, allergic reactions, and gastrointestinal disturbances when consumed excessively (Fong et al., 2021).
Mycotoxin contamination represents another major issue. Although molds are essential for many traditional fermentations, certain species such as Aspergillus flavus may produce aflatoxins under inappropriate environmental conditions. Ethyl carbamate (EC), classified as a probable human carcinogen, can also accumulate during alcoholic fermentation and distillation processes. The coexistence of probiotic functionality and microbial safety concerns highlights the ecological complexity of traditional fermented foods and the need for controlled fermentation strategies (Figure 2). Perhaps more concerning in recent years is the recognition that fermented foods may act as reservoirs for antibiotic resistance genes (ARGs). Anal et al. (2019) emphasized that horizontal gene transfer within fermentation ecosystems could facilitate the spread of resistance determinants into human-associated microbial communities. Although the magnitude of this risk remains under investigation, it underscores the need for improved microbial monitoring and fermentation control.
3.6 Toward Intelligent and Sustainable Fermentation Systems
As demand for traditional fermented foods continues to expand globally, the challenge increasingly lies in preserving artisanal authenticity while ensuring industrial reproducibility and safety. Traditional semi-manual fermentation systems often suffer from inconsistent product quality, contamination risk, and inefficient energy use. Consequently, researchers are now exploring how modern fermentation engineering can support safer and more sustainable production systems.
Jin et al. (2024) proposed that solid-state fermentation (SSF) engineering represents a critical future direction for traditional Chinese fermented food manufacturing. Technologies such as hyperspectral imaging, near-infrared spectroscopy, and automated environmental monitoring allow real-time assessment of fermentation parameters including moisture, acidity, and biomass accumulation. Mathematical modeling further enables prediction of microbial growth kinetics, heat transfer, and metabolite production during industrial scale-up. At the same time, synthetic microbial consortia are emerging as promising tools for precision fermentation. Rather than relying exclusively on uncontrolled spontaneous fermentation, researchers can design microbial communities with targeted functions such as toxin degradation, pathogen inhibition, or enhanced aroma synthesis. Mao et al. (2024) demonstrated that microbial succession strongly influences flavor formation in fermented meat and fish products, suggesting that carefully engineered microbial interactions could preserve traditional sensory characteristics while minimizing safety risks.
Ultimately, the future of traditional Chinese fermented foods likely depends on maintaining this balance between heritage and innovation. Fermentation is not merely a biochemical process; it is also a cultural practice shaped by history, ecology, and human adaptation. As omics technologies continue to uncover the hidden microbial world within these ancient foods, they may provide opportunities not only for safer food production but also for more personalized and microbiome-informed nutritional strategies.
